


Dona ora!

Per un futuro di qualità e un mondo con una cura per la distrofia muscolare di Duchenne e Becker.
Notizie correlate

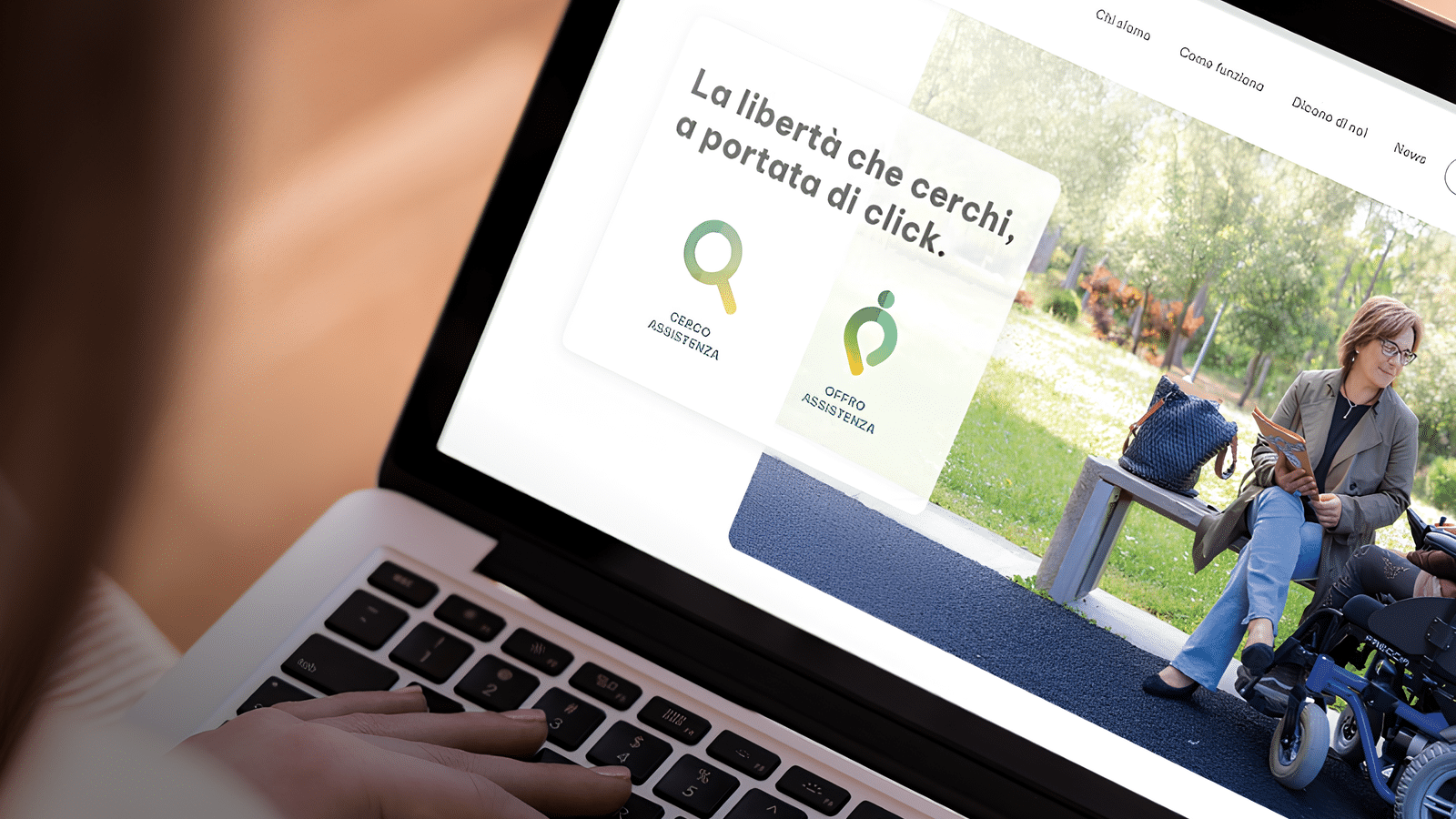
Sostegninrete: la rete dove iniziano storie vere
Da una parte, una persona con disabilità che vuole vivere con più libertà. Dall’altra, un assistente personale pronto a mettere in gioco la propria…

Dyne Therapeutics: risultati positivi dallo studio con z-rostudirsen (Dyne-251) per lo skipping dell’esone 51 nella DMD
In un comunicato stampa diffuso l'8 dicembre 2025, Dyne Therapeutics ha annunciato risultati incoraggianti dallo studio clinico DELIVER di fase 1/2…

Dove inizia la cura: la comunicazione della diagnosi
Il convegno organizzato dall’Università degli Studi Milano-Bicocca e l’associazione Parent Project Sabato 13 dicembre – ore 9 – 17.30 Piazza…
